

Introduction: Type 2 VWD is caused by variants in the von Willebrand factor (VWF) gene leading to impaired function. Distinction between subtypes of Type 2 has traditionally relied on a panel of assays including VWF antigen (VWF:Ag), VWF ristocetin-cofactor activity (VWF:RCo), VWF multimers, and Factor VIII (FVIII:C). In type 2A, there is either impaired synthesis or accelerated degradation of the more adhesive VWF high molecular weight multimers (HMWMs). In type 2B, enhanced VWF-platelet interactions result in binding and removal of the HMWMs, impairing platelet adhesion. Type 2B VWD can manifest spontaneous platelet agglutination and thrombocytopenia. The inheritance patterns of Type 2A and 2B VWD are generally described to be autosomal dominant and fully penetrant.
We identified a multigenerational Canadian First Nations Ojibway kindred with VWD harboring a missense variant in exon 28 (A>T transversion at nucleotide 4898), leading to a single amino acid substitution (p.Asn1633Ile). Some individuals in the kindred are heterozygous, and some homozygous, for the causative variant.
Objectives: The objective of this case series are to describe the genetic basis and pattern of inheritance of this variant of VWD, and to characterize the laboratory hemostasis characteristics in the affected individuals.
Methods: A retrospective case review was conducted on all affected individuals in the kindred known or suspected to harbour this variant. There were 20 individuals seen at our centre over 32 years who were included in this study. We collected clinical and laboratory data to characterize the bleeding phenotype.
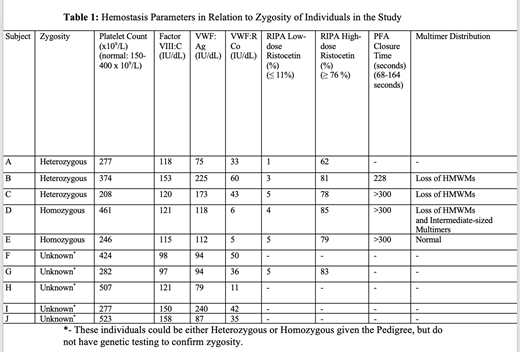
Results: Sixteen of the 20 individuals in the study were known or inferred to be heterozygous, and 4 were homozygous. We were able to examine laboratory hemostasis in ten individuals in the study (Table 1). The platelet count, FVIII:C and VWF:Ag levels were within or slightly above reference range for all individuals. In the heterozygous individuals, VWF:RCo was mildly reduced, VWF multimer testing showed loss of HMWMs, and ristocetin-induced platelet aggregation (RIPA) testing normal aggregation to a higher dose (1.25 mg/ml), a pattern consistent with mild Type 2A VWD (Table 1). In homozygous individuals, VWF:RCo was markedly reduced, and while RIPA was minimal with low-dose ristocetin, aggregation to the higher dose was normal despite the very low ristocetin cofactor activity in the plasma. The PFA-100 (collagen-epinephrine) closure times were markedly prolonged in the homozygous subjects and in one heterozygous subject, exceeding 300 seconds.
Discussion/Conclusion: This novel VWF variant confers a laboratory phenotype consistent with type 2A VWD in the heterozygotes. Homozygotes have more severely impaired hemostatic function. The conventional hallmark of type 2B VWD (hyperresponsiveness to low-dose ristocetin) was not seen, but the fact that full aggregation was obtained in homozygotes despite ristocetin cofactor activities of only 5-6% in the plasma suggests some degree of hyperaggregability, and hence a phenotype more in keeping with Type 2B than 2A. In the future, it would be interesting to see how this missense variant affects GP1b binding to VWF, as RIPA and VWF:RCo are not entirely comparable assays.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal